Human health and disease
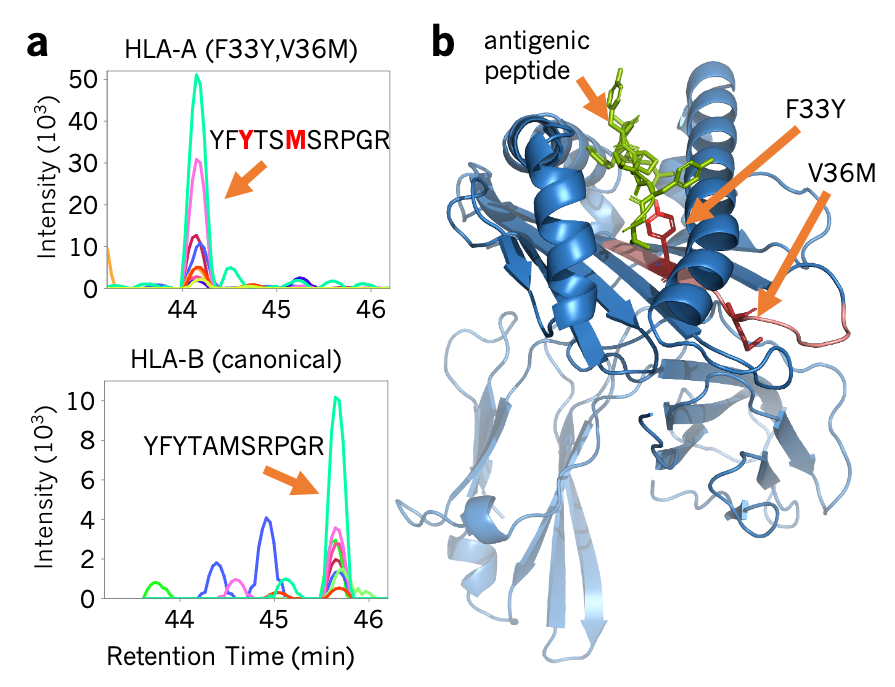
We work on using proteomics to characterize the effects of genetic variation on proteins and protein function, with specific interest in variants that help drive cancer.(a) Fragment ions detected in HLA-A (compared to the same peptides in HLA-B) indicate sequence variants in HeLa. (b) These variants are in the peptide-binding region and likely affect the binding and presentation of antigens to the immune system. Searle et al Nat Comm 2020
Mass spectrometry
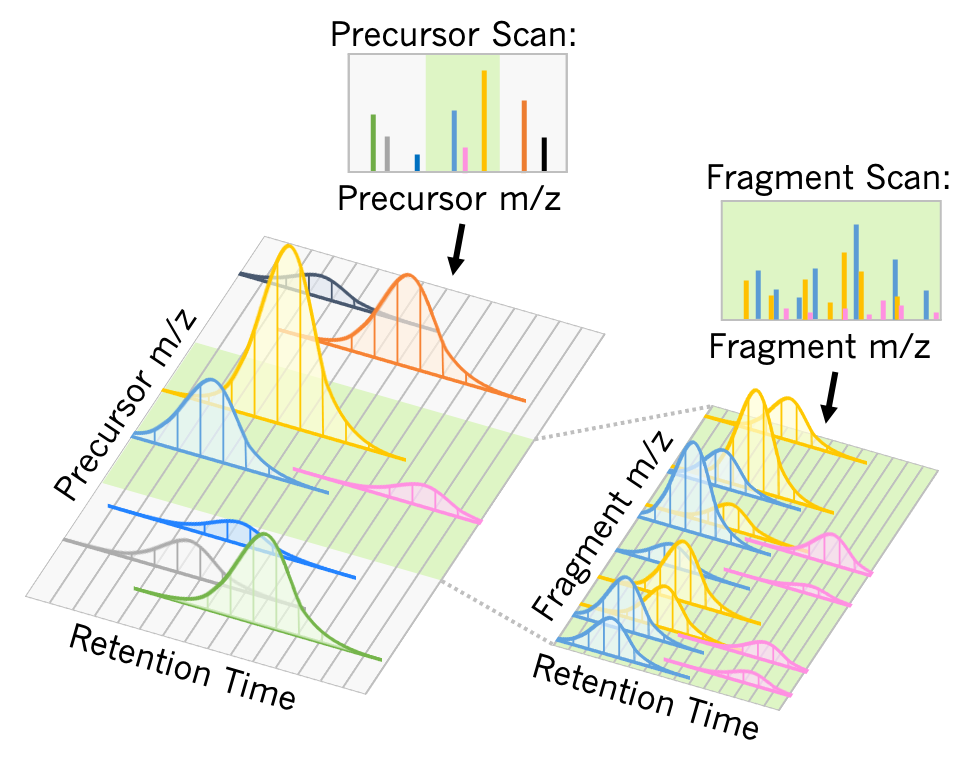
Our lab makes tools for Data Independent Acquisition Mass Spectrometry (DIA-MS). Using DIA-MS, multiple peptides are separated across retention time with liquid chromatography. Peptide precursors in a similar mass range are selected in an isolation window (green highlight) to be co-fragmented. By lining up co-eluting fragment ions across retention time, researchers assign specific fragment ions to individual analytes. MCP tutorial paper by Lindsay Pino and various Youtube videos: (1) , (2) , and (3) .
We use bioinformatics as a key component of all of our work, from developing low-level software for extracting signals from mass spectrometry experiments to building scripts to statistically interpret our biological data. Our focus is on building industry-strength, robust software that broadly enable the research community. You can read more about our current and maintenance supported software projects here . Finally, all software projects we build are released under liberal, open source licenses.
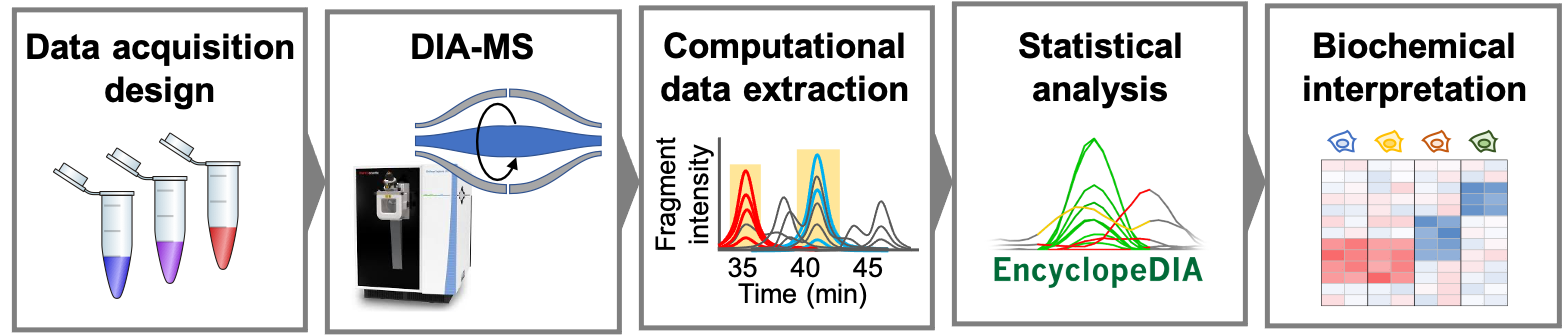 Our lab works on bioinformatics, mass spectrometry, and proteomics technologies as part of the Department of Biomedical Informatics and the Pelotonia Institute for Immuno-Oncology at the Ohio State University. We are interested in developing new methods that improve all aspects of the proteomics workflow, from data acquisition, to computational quantitation and statistical analysis. We are dedicated to open methods and open source software, as well as the long-term support for those computational resources for the proteomics and mass spectrometry communities.
Our lab works on bioinformatics, mass spectrometry, and proteomics technologies as part of the Department of Biomedical Informatics and the Pelotonia Institute for Immuno-Oncology at the Ohio State University. We are interested in developing new methods that improve all aspects of the proteomics workflow, from data acquisition, to computational quantitation and statistical analysis. We are dedicated to open methods and open source software, as well as the long-term support for those computational resources for the proteomics and mass spectrometry communities.